
The CPC research group performs computational (in silico) experiments with atomic resolution based on classical and quantum-mechanical (ab-initio) molecular dynamics simulations. We aim to understand the physical fundamentals of solvation and are especially interested in the unique solvent properties of water. We investigate water-mediated non-covalent interactions that determine the functioning of the living cell and the properties of solutions employed in electrochemical setups and aqueous supramolecular chemistry. To push the boundaries of theoretical chemistry towards quantitative property prediction of complex solutions and soft matter, we are developing force fields, new methods, and software.
Current research in the group revolves around understanding:
- Forcefields and coarse-grained models
- Molecular theory of solutions
- Methods for molecular and multi-scale modelling
- Chemistry and properties of aqueous solutions
- Polymers and soft matter
- Fluids at interfaces
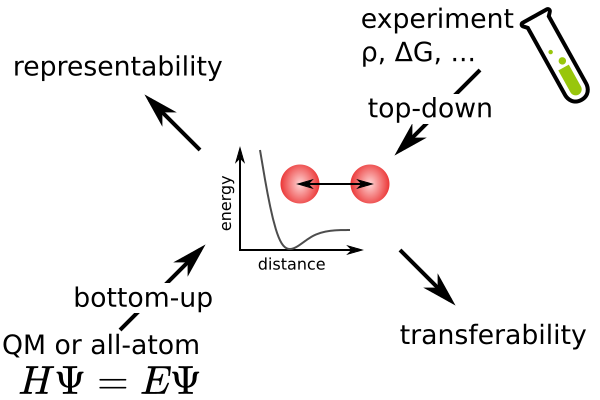
The success of molecular dynamics simulation is based on the force field employed which determines the forces acting on each atom. Its simple form enables fast simulations of complex systems that are far beyond the scope of ab-initio Quantum calculations. The consistency of the underlying forces are very important when using molecular dynamics to analyze emergent properties. Many force fields exist and no single one suits all purposes.
We are working on the generation and improvement of force fields for systems such as polymers and ions in aqueous solutions. Most force fields are derived in a top-down manner and optimized for experimental properties such as free energy of solvation or density. The bottom-up approach is often incorporated to derive partial charges or dihedral potentials from quantum chemistry calculations. We are interested in adapting bottom-up methods to obtain potentials that closely represent the real systems. Bottom-up methods are also in our interest for coarse-grained potentials, that describe the interactions of larger beads representing multiple atoms.
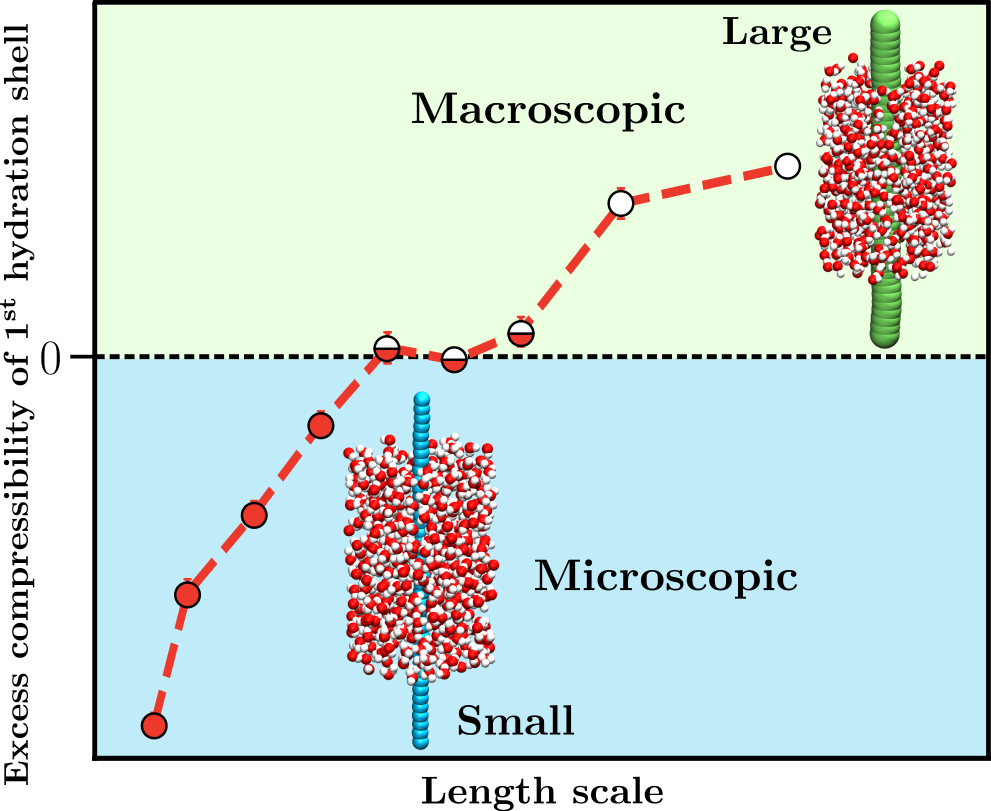
Thermodynamic properties of polymer hydration shells, such as the density fluctuations and compressibility, are crucial in the understanding of polymer coil-globule conformational equilibrium. However, characterizing hydration shell specific properties in computer simulation is not trivial. Toward this, we extend the Small System Method (SSM) to characterize the compressibility of hydration shells of a generic hydrophobic polymer. The SSM exploits the unique feature of finite-sized open systems, whose thermodynamic quantities scale with the inverse system size. This scaling enables the calculation of properties in the thermodynamic limit of macroscopic systems, based on computer simulations of finite-sized systems. We use SSM to study the hydration shell properties of polymers in pure and mixed solvents.
Specifically, we are interested in probing the crossover length scale in polymer hydration and its role in the hydration shell thermodynamics. While the role of a crossover length scale is well known in the case of spherical hydrophobic solutes, the counterpart in the context of hydrophobic polymer has not been investigated. Subsequently, we want to investigate these properties in the presence of various co-solvents in order to understand their specific roles.
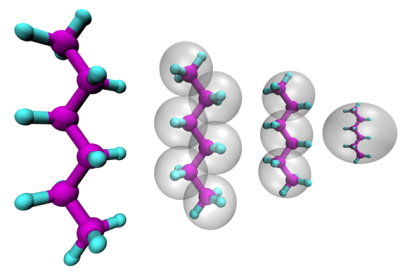
The properties of soft-matter materials depend on the structure and dynamics on a very wide range of time and length scales. On the smallest scale quantum-chemical calculations are routinely used to determine the structure of small molecules and can be also utilized to study the dynamics of a small set of molecules with ab-initio molecular dynamics simulations. The application of these methods is typically limited to small system sizes and/or short trajectories. Classical molecular dynamics simulations rely on empirical force-fields which have to be carefully chosen to correctly represent the system of interest. Given a high-quality force field this method allows to simulate thousands of atoms and to reach time scales nano-seconds while still preserving atomistic detail. On the macroscopic scale continuum approaches can be utilized with the downside of loosing all information of molecular processes.
To bridge the scales between atomistic and continuum resolution we develop and apply coarse-graining techniques with which the number of degrees of freedom is reduced from an atomistic representation to a representation based larger pseudo-atoms. The challenge is to derive models in a bottom-up approach which accurately represent an underlying high resolution model, but allow for a significantly increased computational efficiency. The close linkage of bottom-up derived coarse-grained models to its underlying fine-grained counterpart allows a simultaneous analysis on a wider range of length and time scales with models, which by construction consistently describe the same physical systems
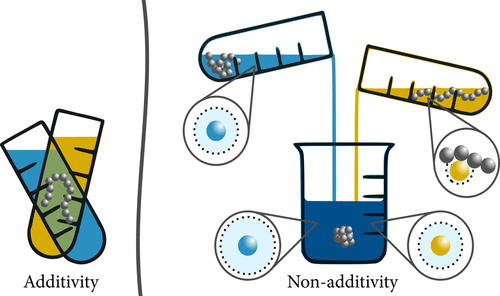
Responsive polymer materials undergo changes in material properties in the presence of external stimuli such as temperature, pH, cosolvent (cosolutes) concentration. This stimuli responsive behavior is at the heart of applications such as drug delivery, tissue engineering, smart coatings and bio-sensors. We use a combination of atomistic molecular dynamics simulations and free energy methods to understand the mechanisms driving such phenomena in polymer solutions with cosolvents such as alcohols, acetone and cosolutes such a urea, TMAO and salts. These systems exhibit a wide variety of responsive behavior which depend on aspects such as preferential adsorption, excluded volume interactions and polymer-solvent cohesive interactions. Interestingly, “Non-additive behavior” is a norm rather than the exception in such systems.
Example
Cononsolvency is the phenomenon where polymers undergo coil-globule-coil transitions in binary mixtures of two good solvents. Poly(N-isopropylacrylamide) and Poly(N,N-diethyl acrylamide) are well known polymers which exhibit such transitions with addition of cosolvents such as alcohols, acetone, dimethylsulfoxide and dimethyl formamide. Our group aims to understand the mechanism driving this phenomenon through different aspects. One aspect is to improve existing atomistic forcefields in order to match the simulation results with experiments. On the other hand, we also employ model polymer systems to understand the role different contributions such as preferential adsorption, excluded volume interactions and polymer-solvent/cosolvent attraction. Recent work from our group shows that alcohols, through a surfactant mechanism, favor polymer collapse even though they reduce the solvation free energy of the hydrophobic polymer-solvent interface.
Macromolecules 2019, 52, 11, 4131–4138
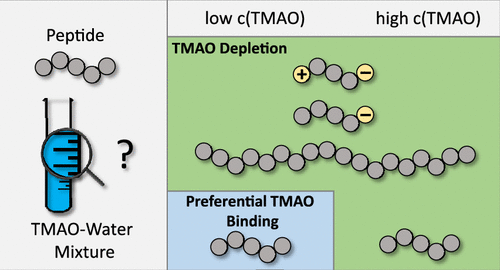
Pressure, Peptides, and a Piezolyte: Trimethylamine-N-oxide (TMAO) is a well known osmolyte counteract the denaturation of proteins due to hydrostatic pressure in biological systems. The role of TMAO stands out since the amount of TMAO in muscle tissue of deep sea fish increases with the depth of the catch and thus with higher pressure. Therefore, TMAO is often called a piezolyte due to its ability to counteract detrimental pressure effects on proteins. Our group, through a combination of experiments and simulations, aims to understand the interaction of TMAO with peptides and its dependence on aspects such as pressure, peptide chemistry and TMAO concentration.
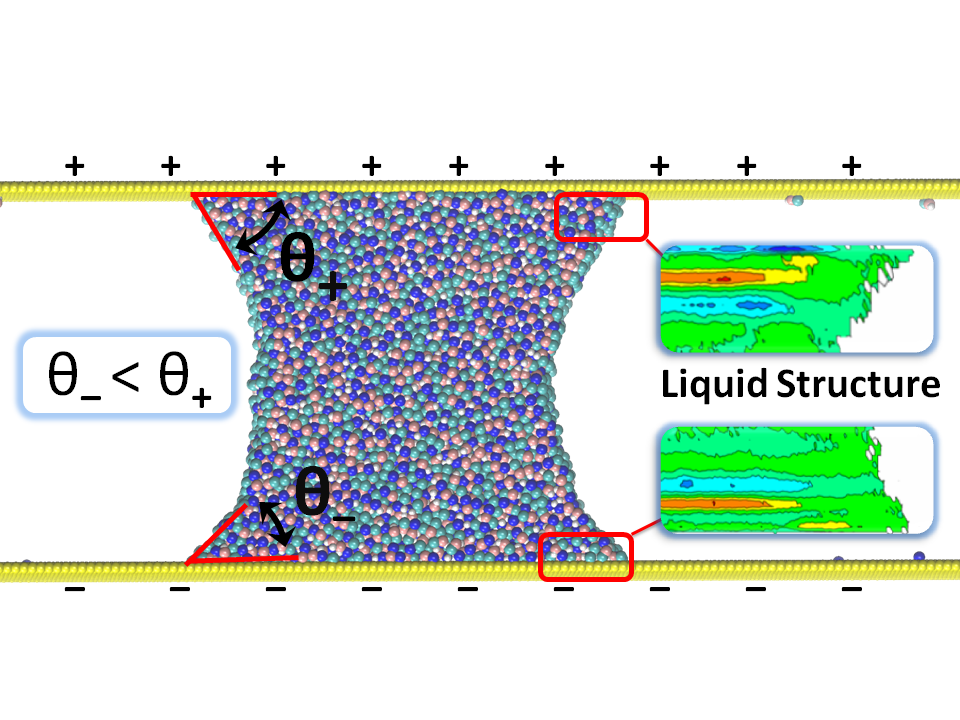
Several elements impact the structural behavior of a liquid on a surface and its dissipation near the interface, the most important of which are surface tensions, liquid-solid interaction, and the roughness of the solid surface. We perform atomistic and coarse grained simulations to further understand this relationship.
Our major focus of research in recent years has been the study and development of methods to compute interfacial friction at the solid-liquid interface using equilibrium fluctuations. We have also been concentrating on expanding these approaches to better understand wetting phenomena on soft and deformable surfaces, such as polymer surfaces.